Open Source Infrastructure for Differentiable Density Functional Theory
Deep Forest Sciences
Rida Irfan, Advika Vidyadhiraja, Bharath Ramsundar
01.10.2024
Understanding Density Functional Theory
Density-Functional Theory (DFT) is a cornerstone of quantum chemistry that provides a more efficient method to understand the electronic structure of atoms, molecules, and materials. DFT simplifies the complex Schrödinger equation for many-electron systems by replacing the intricate many-body wave function with an electron density function which depends only on three spatial coordinates. This substitution allows us to describe the electronic ground state of complex molecular systems through single-particle equations and an effective potential comprised of three key components,
- The Exchange-Correlation (XC) Potential, accounting for many-body effects.
- The Hartree Potential, describing the electrostatic electron-electron interaction.
- The Ionic Potential, resulting from the atomic cores.
In this blog post, we discuss our recent article accepted into ICML'2023 Synergy and Scientific and Machine Learning Modeling (SynS & ML) workshop, and discuss our open-source model for Differentiable DFT which leverages machine learning to learn a predictive exchange correlation functionals. This work aligns with the broader efforts of Deep Forest Sciences, built around the open-source DeepChemecosystem, to leverage powerful machine learning tools (including large chemical foundation models like ChemBERTa and ChemBERTa-2) to accelerate small molecule drug discovery. The integration of DeepChem with DFT could have significant implications for the field of drug discovery by helping us improve covalent docking and retrosynthesis.
DeepChem and DFT
To make differentiable DFT more efficient and accessible, we've developed open-source infrastructure aimed at training neural exchange correlation functionals. Our aim is to standardize and streamline the process of learning these functionals. We have open-sourced the model in DeepChem, an open-source Python library designed to facilitate scientific machine learning calculations by breaking them down into structured workflows. DeepChem is tightly integrated into the Deep Forest Sciences ecosystem.
The DFT community has started moving towards differentiable DFT due to challenges with defining XC functionals for complex systems. This 2021 paper from Oxford presents a model, XCNN, that leverages deep neural networks to learn XC functionals and combine them with traditional XC functionals. This innovative approach is not system-specific, allowing it to be applied to a broad spectrum of physical systems. Another recent advancement is the development of the DM21 functional by DeepMind, computed using fictional systems with fractional charge and spin constraints in order to overcome errors encountered by traditional XC functionals. DM21's method is particularly valuable when dealing with charge densities involving mobile charges and spins.
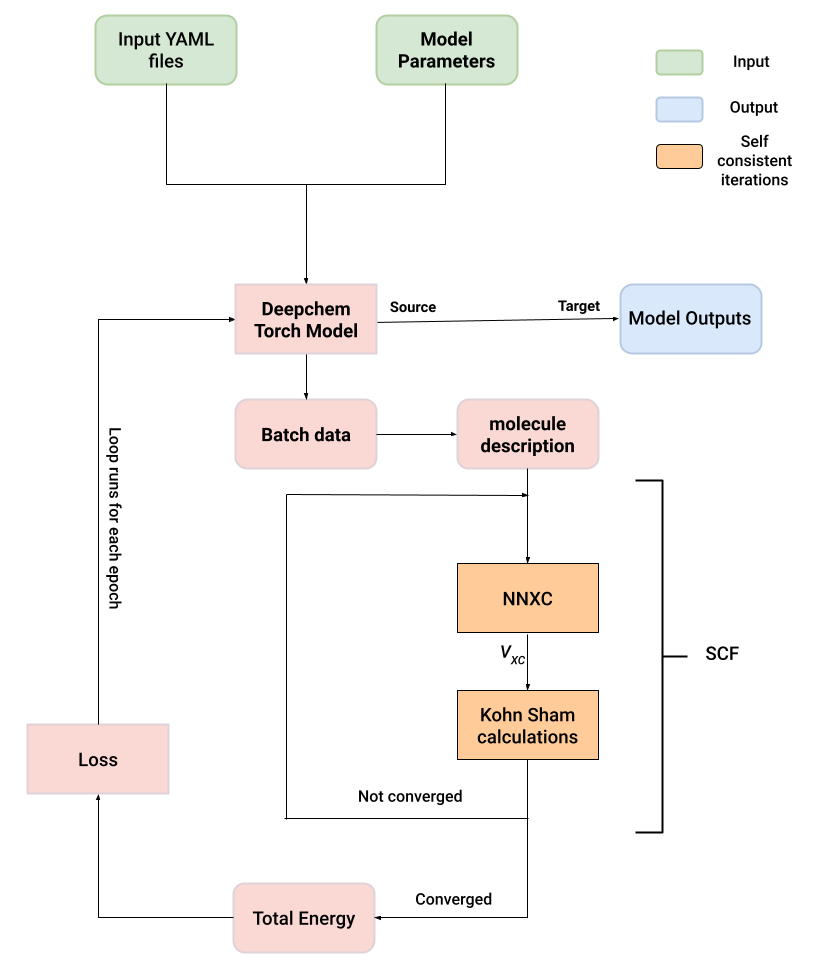
We aim to standardize differentiable DFT workflows, by making the process of training neural network XC functionals more accessible through DeepChem’s infrastructure. This standardization retains model accuracy, and is validated through experiments and comparisons with the original XCNN model. The XCModel, based on the XCNN implementation in DeepChem (adapted and modified from Oxford’s open source release), provides flexibility by including different loss functions, metrics, and machine learning models present in DeepChem. This infrastructure will facilitate the incorporation of more comprehensive density information and fractional constraints akin to the DM21 functional from DeepMind and will help foster the development of new hybrid functionals trained using both neural networks and a fully differentiable quantum chemistry library. The diagram below shows an architectural schematic of DeepChem’s differentiable DFT infrastructure.
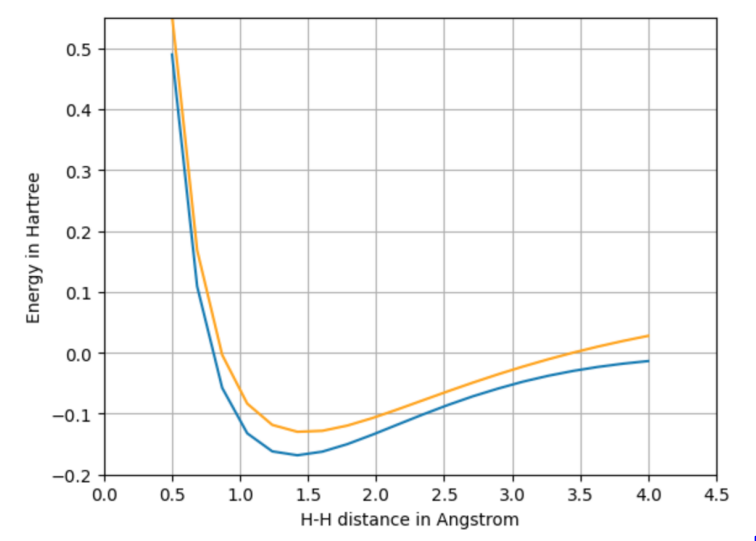
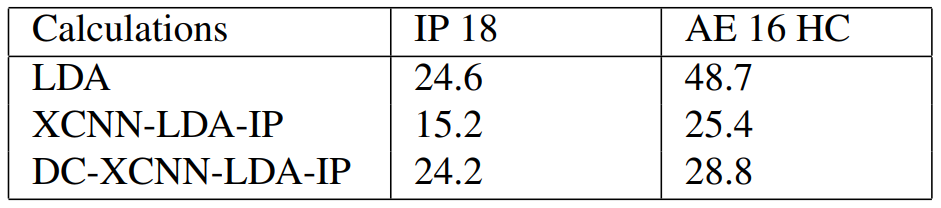
We tested our implementation by computing a hydrogen dissociation curve as shown in the figure below. In this experiment, we compare Mean Absolute Error (MAE) values for ionization potential and atomization energy using different functionals. Although our DeepChem implementation slightly trails the original, we expect to close this gap with further refinement. The experiment was executed on a 16GB RAM CPU, and illustrates how the XCModel accurately tracks dissociation energy, indicating its potential for precision in future larger-scale simulations.
Conclusion
Open-source infrastructure for differentiable DFT in DeepChem can help simplify complex calculations, foster collaboration, and improve accuracy . Our research aligns with Deep Forest Sciences’ broader aim to accelerate AI-driven drug discovery. Future developments, including fractional constraints on fictional systems and Meta-PBE layers, can make the model even more versatile and accurate, especially for future applications in drug discovery such as covalent inhibitor design.
Deep Forest Sciences Prithvi suite provides a rich toolkit of computational approaches for drug discovery. Get in touch if you would like to use our cutting-edge AI approaches to accelerate your small molecule discovery efforts today!
About Deep Forest Sciences
Deep Forest Sciences’ AI-powered Prithvi toolchain accelerates small-molecule drug discovery efforts. Deep Forest Sciences also leads the development of open-source DeepChem framework, and emphasizes supporting open source and open science as fundamental parts of our mission and values. Partner with us to apply our foundational AI technologies to hard real-world problems in small molecule drug discovery.
Email us at partnerships@deepforestsci.com!