No-Code Molecular Docking with Prithvi
Deep Forest Sciences
Jose Antonio Siguenza, Bharath Ramsundar
04.09.2024
A Brief Introduction to Molecular Docking
Molecular docking is a computational method that predicts how small molecules, with atomic weight less than ~500 dalton, bind to larger biomolecular targets like proteins. Docking roughly simulates interactions between molecules to determine favorable binding positions for ligands to proteins and is utilized in drug discovery for screening and understanding molecular interactions.
A Brief Introduction to Prithvi
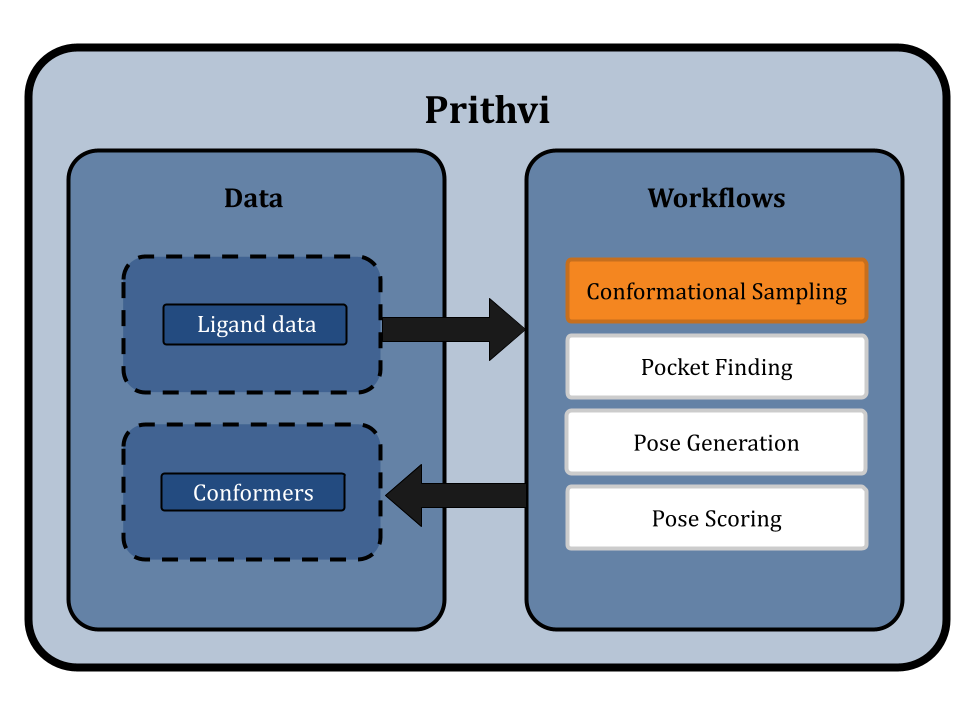
Prithvi is our AI-powered platform to accelerate small molecule drug discovery with scientific foundation models. Prithvi has the ability to
- Perform structural analysis of targets and identify potential binding sites for a more targeted design process.
- Build models based on early assay or patent data for use in large-scale virtual screens.
- Construct active learning pipelines to identify and confirm high-quality hits.
- Guide retrosynthetic analysis.
- Suggest modifications to increase the potency, selectivity, and safety of hits.
To learn more about Prithvi, check out our earlier blog post. In this post, we will focus on using Prithvi for molecular docking analyses, but in future posts we will highlight Prithvi’s capabilities in free energy calculations, molecular dynamics, and more.
Prior Context about Molecular Docking
Proteins or enzymes related to biological processes or disease pathways are the central targets of modern drug discovery. Drug discoverers seek to design compounds, or ligands, that interact with these targets in various ways. A binding site (or pocket) is a region of the target where a drug-like molecule can effectively interact with it. The drug-target pair will tend to occupy low energy states since those are more physically favorable. A particular choice of drug-target interaction shape is called a binding pose. The shape of the molecule in this pose is called a conformation. Molecular docking aims to predict low-energy ligand conformations associated with high-affinity binding sites of targets. Additional biomolecules, like DNA, can also be involved when drugging systems like transcription factors.
Several families of classical algorithms predict low-energy binding affinity using score functions based on physical or chemical equations [1]. Ligand chirality, Van der Waals forces, electrostatics, and hydrophobicity are important in calculating binding affinity and are often explicitly included in physical scoring functions. However, such methods often have high runtime and associated computational costs. Recent advances in deep learning have enabled algorithms to learn from molecular data and enhance pose search space while reducing the parameters mentioned earlier, but possibly at the cost of failing to generalize to proteins not present in the training dataset.
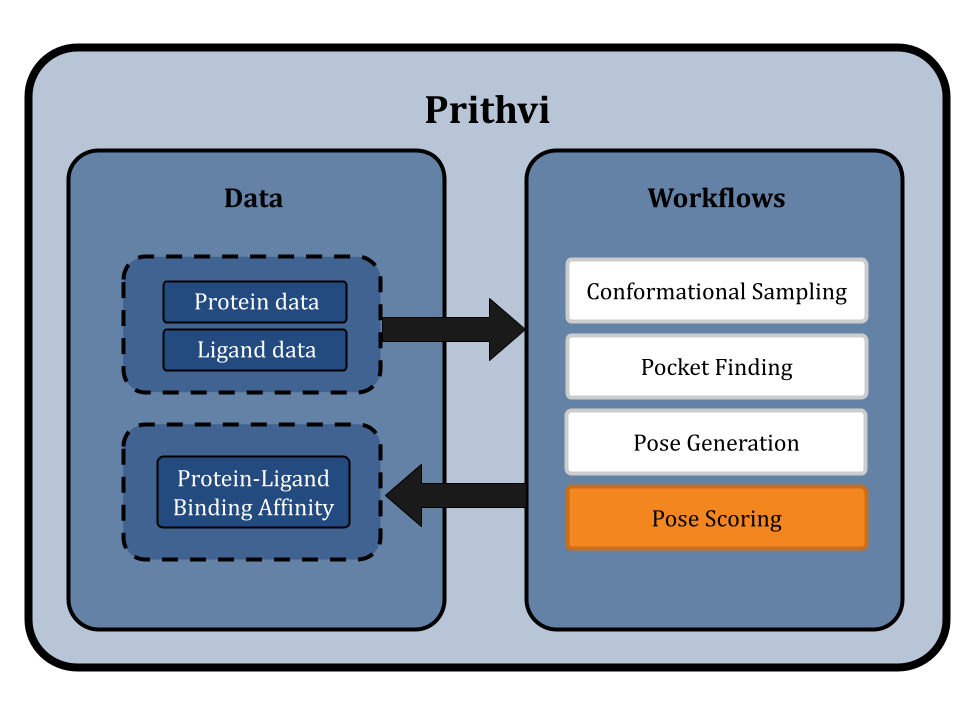
Molecular Docking with Prithvi
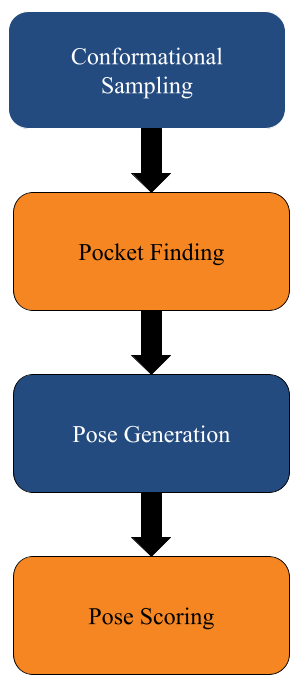
This blog post highlights Prithvi’s molecular docking features to explore protein-ligand interactions. Prithvi provides a protein-handling infrastructure suite to run docking jobs. This tool broadens a company’s molecular search space, and helps identify potential drug-like molecules. Below is a general workflow to perform a docking simulation on Prithvi:
- PDB Cleaning
- Conformational Sampling
- Pocket Finding
- Pose Generation
- Pose Scoring
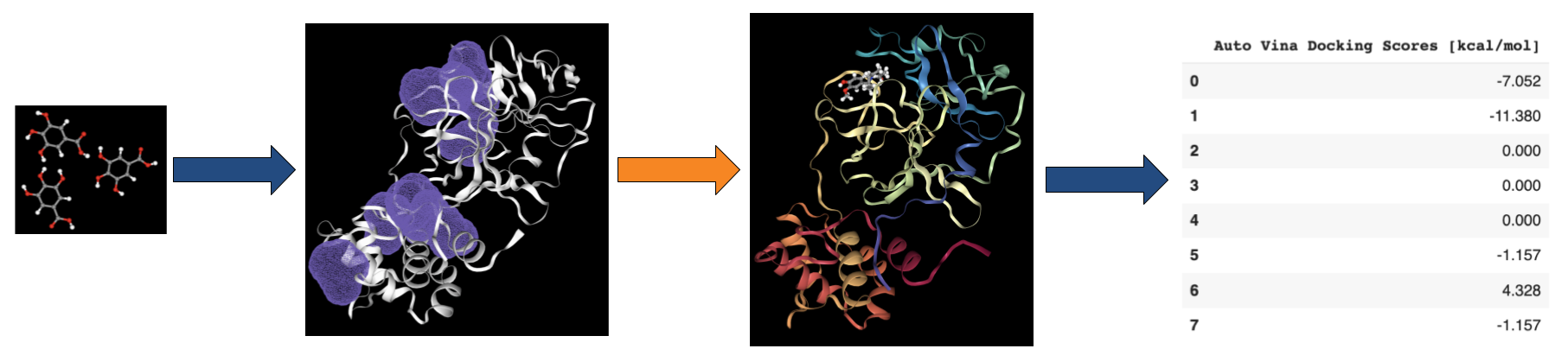
Prithvi uses open-source tools like DeepChem and AutoDock Vina to perform docking. Open-source tools enable us to apply state-of-the-art scientific tooling maintained by a broader community. Prithvi’s no-code infrastructure makes advanced computational methods easily accessible to biologists and chemists. In this blog post, we perform a case study to showcase Prithvi’s no-code molecular docking capabilities by docking five organic compounds (Naringin, Capsaicin, Gallic Acid, Psychotrine, and Quercetin) against the SARS-CoV-2 main protease (PDB ID:5R80) [2].
Each role outlined in Prithvi’s docking workflow is described below.
Conformational Sampling: Sample Low-Energy 3D Molecular Shapes
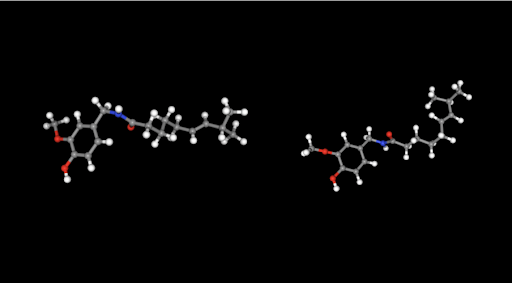
Docking aims to select a low-energy binding pose among different binding poses. A molecular conformation is one of the plausible spatial arrangements of atoms in a molecule. Conformer sampling is based on axis rotations and atomic bond restrictions [6]. Therefore, the ligand’s structure limits the number of conformers. Larger molecules generate more conformers due to increased degrees of freedom (e.g., rotation modes). Ligands are normally input into Prithvi as SDF, PDB, or SMILE files. If desired molecules aren’t known upfront, scientists can use Prithvi’s built-in editor tools to draw the molecular structure and download it as the previous file types. Pairing docking algorithms with good conformational sampling explores a broader ligand space, and increases chances of identifying native-like binding poses increase.
Prithvi’s conformational sampling primitive asks for an input file, containing a ligand, the number of conformers to generate and the output filename to store the sampled conformation. Prithvi currently computes the results by using RDKit to generate conformers, but will soon support more powerful sampling methods.
Finding Binding Pockets: Mapping the Topography of Molecular Interaction Sites
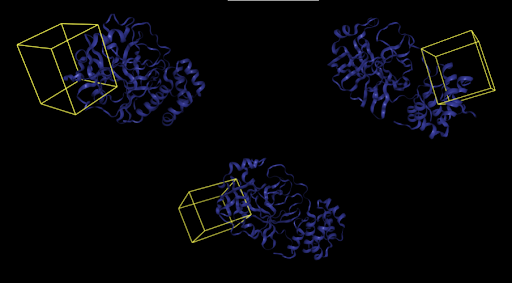
Protein-ligand interactions take place at preferred sites on the target protein commonly referred to as binding pockets. Docking can be performed with or without knowledge of the binding pocket. Docking performed without knowledge of a binding pocket is referred to as blind docking, in contrast to docking performed with a specified binding pocket which is non-blind. Non-blind docking is usually more efficient but in some cases a good binding pocket is not known.
In blind docking, the whole protein surface must be searched for possible binding pockets. In contrast, with non-blind docking users can specify bounding boxes as parameters, strategically defining spaces where a ligand is most likely to bind. Users can visualize these bounding boxes directly through Prithvi.
Several methods have been proposed to detect binding pockets including geometry-based, energy-based, sequence-based, hybrid, and deep learning algorithms [3]. Different algorithms can be more or less effective for different systems.
Prithvi supports a geometric-based approach based on DeepChem’s docking utilities. This method computes the convex hull of the protein and uses it to identify faces that may be suitable for docking. More advanced strategies that leverage Voronoi tessellations and deep learning-based surface scanning methods are under development and will be added to Prithvi shortly.
Pose Generation: The Molecular Arrangement
After exploring the conformer space and binding sites, the next step is coupling the ligand to the receptor. Molecular docking aims to predict protein-ligand binding mode candidates, known as poses [1]. Based on user-defined parameters, a ligand is assessed at different potential binding pockets on the protein surface, and favorable poses are sampled (or generated in the case of deep methods). The poses with lowest binding energy have greater energetic stability.
The pose generation primitive in Prithvi currently uses AutoDock Vina and Gnina as its docking engines [4]. Parameters must be specified for the ligand, the protein to dock against, exhaustiveness, and the number of poses to generate. Centroid and box dimension parameters can optionally be provided to perform non-blind docking. We are also working on incorporating new deep-learning and equivariant pose generation methods into our pipeline.
Pose Scoring: Quantitative Assessment of Molecular Affinity
Pose scoring quantifies energetically favorable binding poses by estimating the strength of the protein-ligand binding interaction. As mentioned previously, this interaction depends on chemical and physical factors such as Van der Waals forces, electrostatic interactions, and hydrogen bonding which contribute to the numerical score, typically calculated in kcal/mol [1]. Docking methods aim to predict pose orientation and stability by comparing binding energies. For Autodock Vina, this optimization task involves genetic algorithms iterating over search space to identify favorable conformers with low binding energy.
Prithvi’s pose scoring primitive needs a docked ligand, a target as inputs, and a filename to store results as a CSV file. Audock Vina, based on simple physical approximation, or Gnina’s score function, based on a convolutional neural network, can be used to compute a binding score [5].

A Case Study with Prithvi’s No-Code Docking
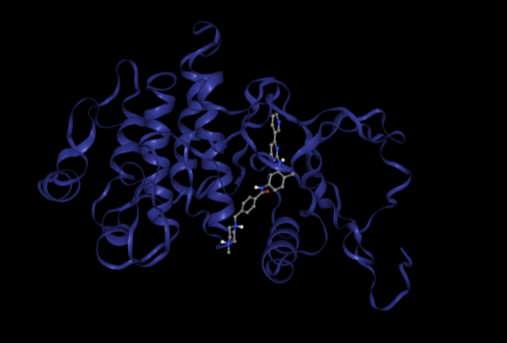
We start the case study with a control experiment to evaluate Prithvi’s performance on a known benchmark system. Autodock Vina provides an example that docks imatinib with the kinase domain of c-Abl (PDB:1IEP) [7] (link). We run the same experiment with Prithvi and display below the scores from Prithvi’s no-code Vina docking and the reference Autodock Vina scores. Files 1iep and imatinib were retrieved from Autodock Vina’s repository.
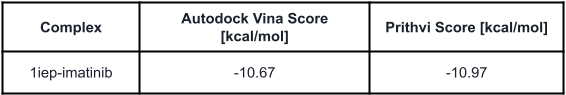
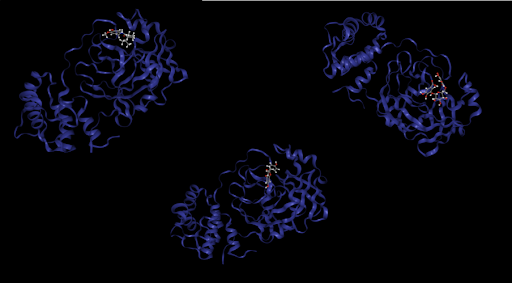
Next, we study some potential inhibitors for the SARS-CoV-2 main protease [2]. Active components from medicinal plants have shown beneficial effects such as antioxidant, antidiabetic, or anti-inflammatory activity with good toxicity profiles. Consequently, following [2], we choose the following compounds for docking against the main protease:
- Capsaicin
- Gallic Acid
- Quercetin
- Psychotrine
- Naringin
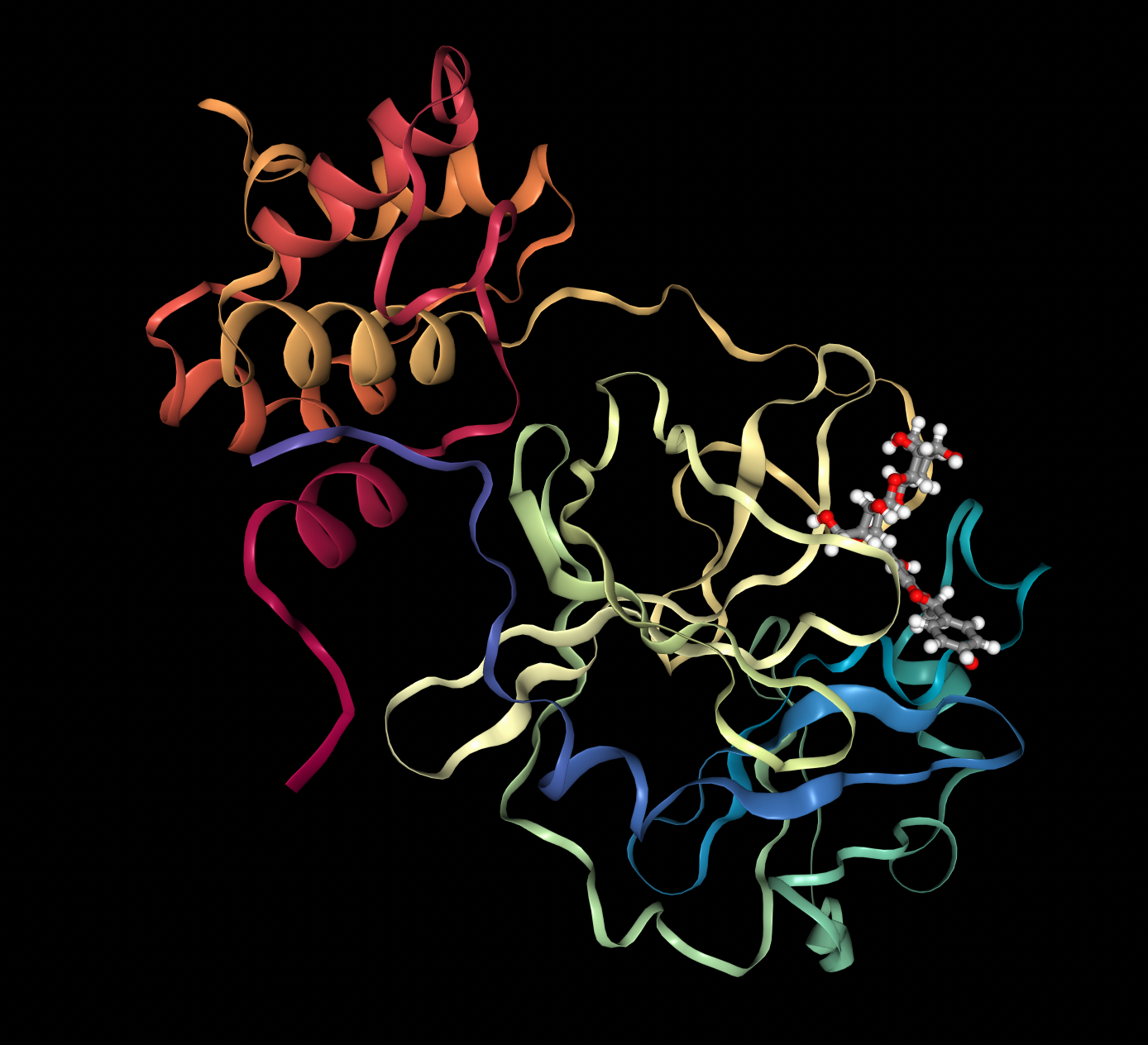
Prithvi offers an easy-to-use UI to visualize conformations and binding poses. The images below were rendered using Prtihvi’s visualization features. Below are five organic compounds and a SARS-CoV-2 main protease co-crystal structure.
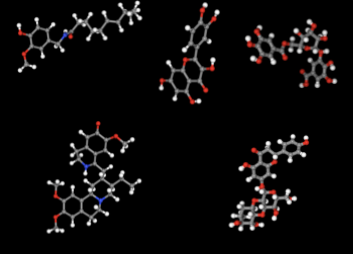
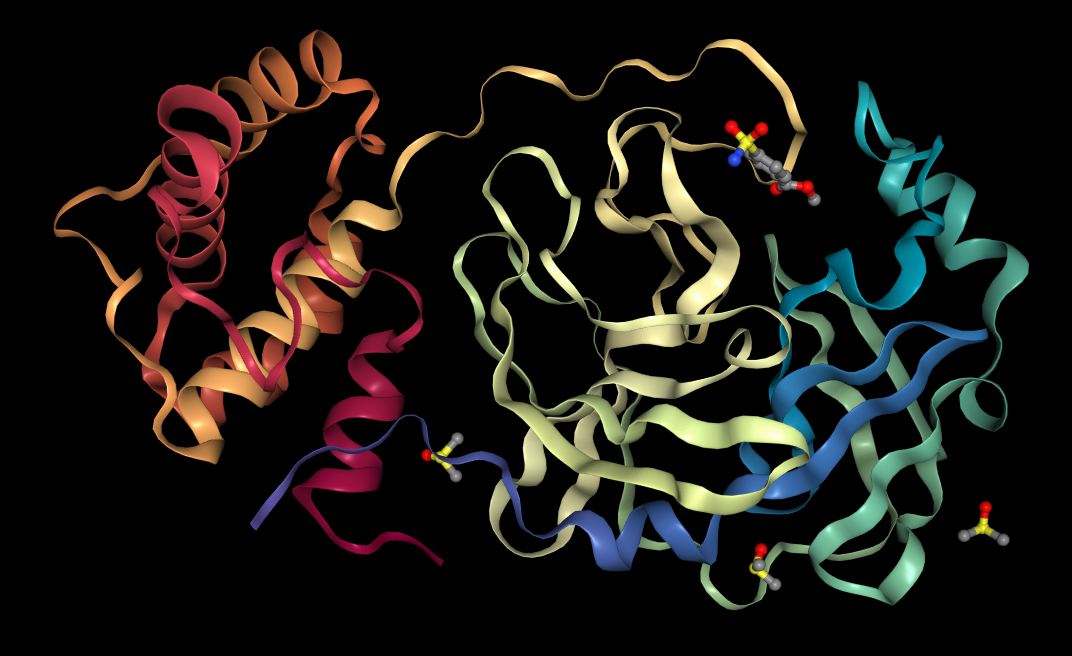
First, 5 conformers were sampled for each ligand. Then, the pocket-finding primitive was used to identify possible binding sites on the SARS-CoV-2 main protease. Visualizing binding pockets can help support or discard hypotheses about where drug candidates may bind. Below, a binding pocket is presented for the main protease co-crystal structure where molecular fragments are enclosed [2].
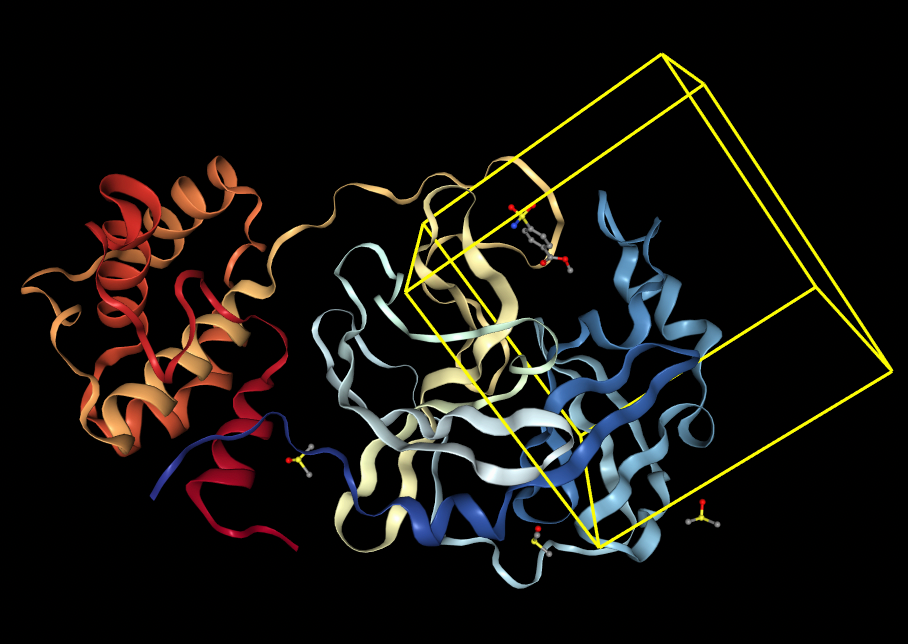
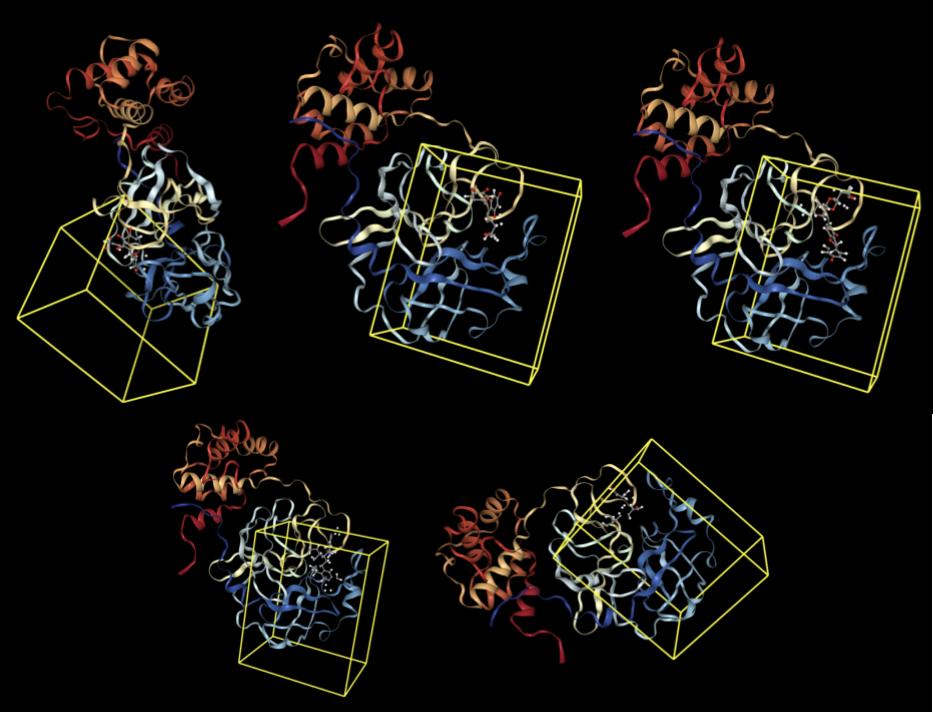
The pose generation primitive parameters were set to output 2 poses for each ligand conformer. Below are presented protein-ligand complexes of each ligand and SARS-CoV-2 main protease. Poses are likely to bind inside a bounding box colored yellow retrieved from Pocket Finding primitive. This structure is the same as above and serves as a guide to proceed with docking.
Results from Prithvi’s score primitive are presented below:

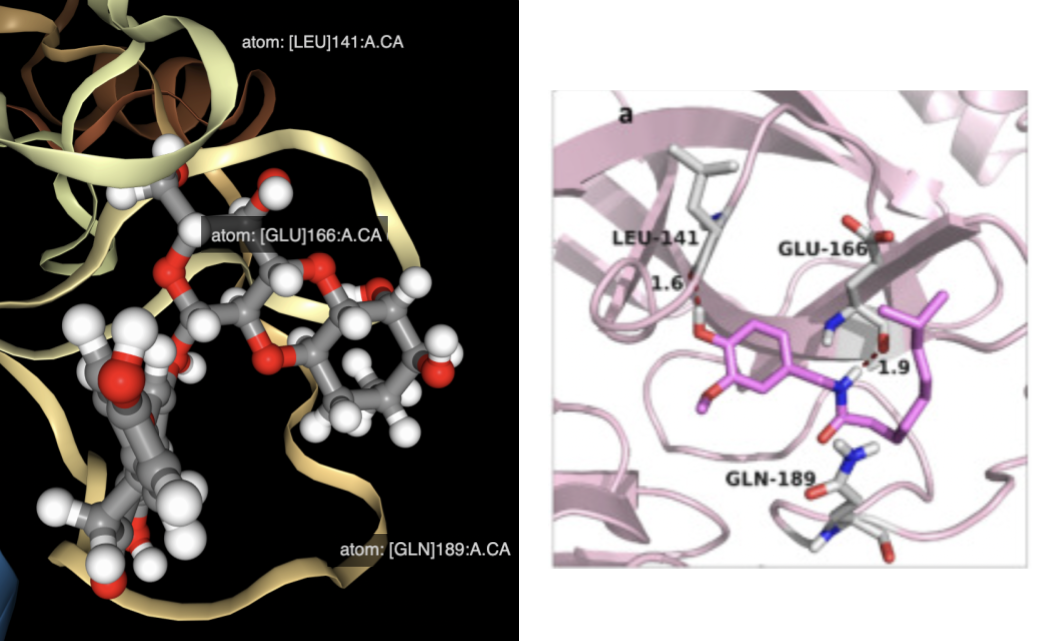
Naringin presented the lowest score compared with other candidates. Naringin’s lowest energy binding pose occupies a binding site formed by LEU-141, GLU-166, and GLN-189 residues, matching results from the reference study [2].
Conclusions and Takeaways
Prithvi provides a powerful no-code framework to execute complex drug discovery workflows. This blog post showcases Prithvi’s no-code molecular docking workflows and capabilities. Biologists and chemists can leverage Prithvi’s powerful UI to run complex analyses without having to write custom scripts. Prithvi also supports very-large scale docking workflows. In a future blog post, we will discuss how to run very-large scale docking using Prithvi.
About Deep Forest Sciences
Deep Forest Sciences’ AI-powered Prithvi toolchain accelerates small-molecule drug discovery efforts. Deep Forest Sciences also leads the development of open-source DeepChem framework, and emphasizes supporting open source and open science as fundamental parts of our mission and values. Partner with us to apply our foundational AI technologies to hard real-world problems in small molecule drug discovery.
Email us at partnerships@deepforestsci.com
References
- R. Meli, G. M. Morris, and P. C. Biggin, “Scoring Functions for Protein-Ligand Binding Affinity Prediction Using Structure-based Deep Learning: A Review,” Front. Bioinforma., vol. 2, p. 885983, Jun. 2022, doi: 10.3389/FBINF.2022.885983/BIBTEX.
- A. A. Alrasheid, M. Y. Babiker, and T. A. Awad, “Evaluation of certain medicinal plants compounds as new potential inhibitors of novel corona virus (COVID-19) using molecular docking analysis,” Silico Pharmacol., vol. 9, no. 1, pp. 1–7, 2021, doi: 10.1007/s40203-020-00073-8.
- T. Kawabata, “Detection of multiscale pockets on protein surfaces using mathematical morphology,” Proteins Struct. Funct. Bioinforma., vol. 78, no. 5, pp. 1195–1211, 2010, doi: 10.1002/prot.22639.
- J. Eberhardt, D. Santos-Martins, A. F. Tillack, and S. Forli, “AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings,” J. Chem. Inf. Model., vol. 61, no. 8, pp. 3891–3898, 2021, doi: 10.1021/acs.jcim.1c00203.
- C. Shen, J. Ding, Z. Wang, D. Cao, X. Ding, and T. Hou, “From machine learning to deep learning: Advances in scoring functions for protein–ligand docking,” Wiley Interdiscip. Rev. Comput. Mol. Sci., vol. 10, no. 1, pp. 1–23, 2020, doi: 10.1002/wcms.1429.
- J. Shim and A. D. MacKerell, “Computational ligand-based rational design: Role of conformational sampling and force fields in model development,” Medchemcomm, vol. 2, no. 5, pp. 356–370, 2011, doi: 10.1039/c1md00044f.
- B. Nagar et al., “Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571),” Cancer Res., vol. 62, no. 15, pp. 4236–4243, 2002.