No-Code Relative Binding Free Energy (RBFE) Calculations with Prithvi
Deep Forest Sciences
Dibyam Kumar, Rida Irfan, Bharath Ramsundar
07.15.2024
A Brief Overview of RBFE
For many years, accurate in-silico prediction of protein-ligand binding affinities has been a fundamental goal in computational drug design, due to its potential to accelerate the discovery process. However, traditional computational methods have often failed to provide accurate results. Recent advances have improved the state of the art in Relative Binding Free Energy (RBFE) calculations, which use physics-based molecular simulations and statistical mechanics, to the point where accurate protein-ligand binding affinity prediction is now within sight.
Taking a step back, RBFE calculations are computational methods used in the field of computational chemistry and drug discovery to estimate the difference in binding free energy between two or more ligands to a common target (e.g., a protein or enzyme). Binding free energy is a key factor in understanding and predicting the affinity of a ligand for its target, which is crucial in drug design.
RBFE Calculations
The binding free energy is a thermodynamic parameter that represents the balance between the energy gained from the favorable interactions between the ligand and the target (e.g., van der Waals forces, hydrogen bonding) and the energy cost associated with desolvation and conformational changes upon binding. RBFE calculations involve comparing the binding free energies of different ligands and determining the differences between them. The goal is often to rank different ligands based on their binding affinities or to understand the factors contributing to the binding affinity differences.
Examining the thermodynamic cycle at hand, our focus will be on finding the change in binding energy between molecules A and B with respect to a given protein target.
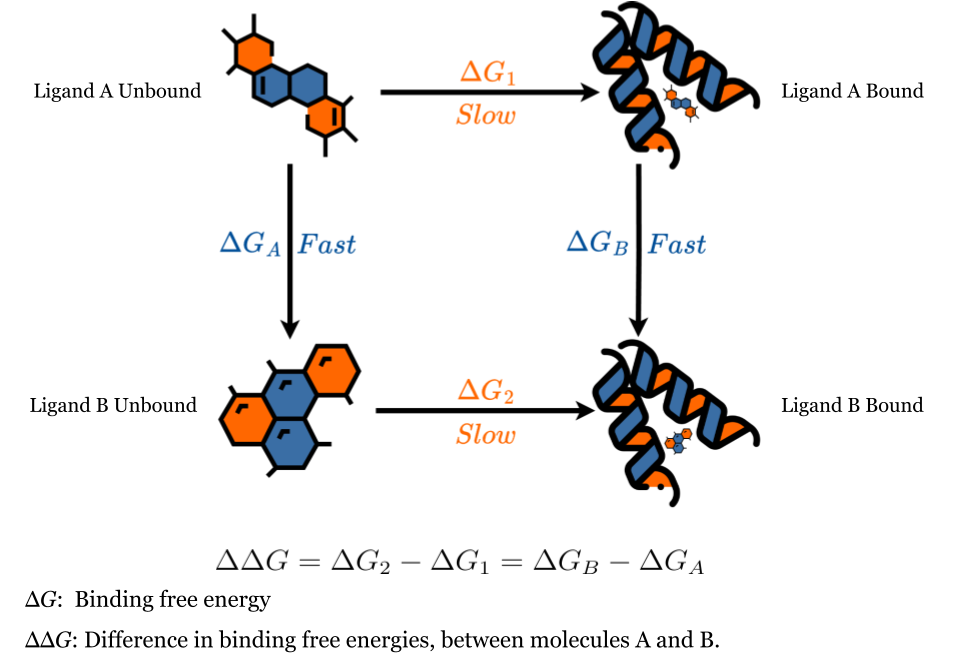
Two methodologies exist for computing the difference in binding free energy, between molecules A and B. The direct method, depicted by the horizontal lines above, involves a direct transition from the bound state of A to the unbound state of B. This technique exhibits slow convergence attributed to the substantial conformational difference between initial and final states. Another approach is the “alchemical” path, depicted by vertical lines, which incorporates gradual modifications between the states. This method exhibits faster convergence by implementing smaller, gradual transitions. In this strategy, molecule A undergoes a gradual transformation (or perturbation) into molecule B in both the bound and unbound states (vertical lines). The difference in free energy between these transformations equates to that of the direct method, owing to the closed thermodynamic cycle.
No-Code Relative Binding Free Energy Calculations with Prithvi
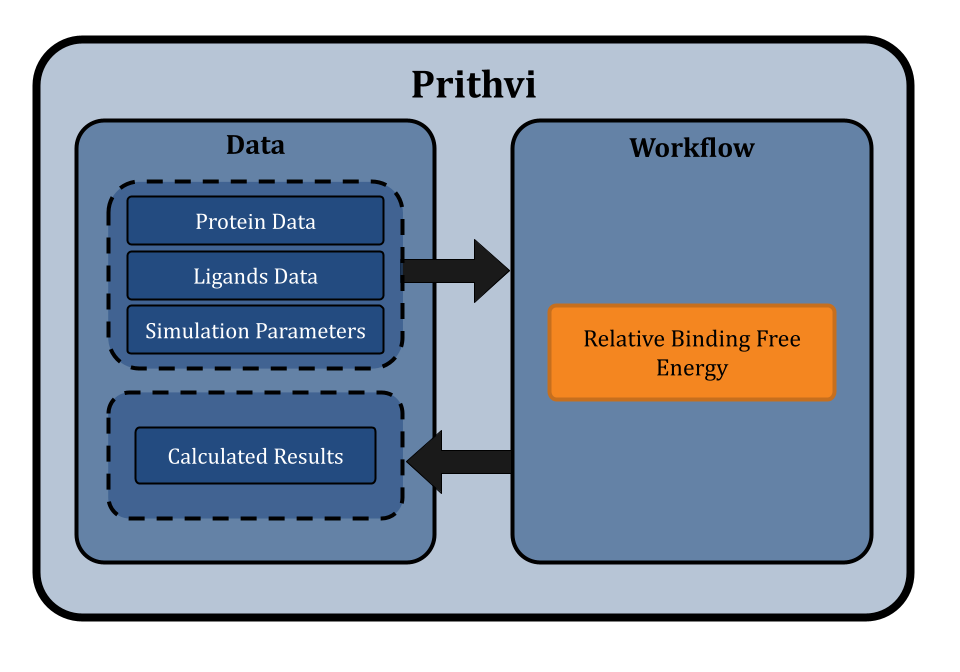
Relative binding free energy calculations are typically complex to set up and require significant expertise to prepare and analyze results. To address this challenge, Deep Forest Sciences has built no-code infrastructure on the Prithvi™ platform for researchers to run RBFE calculations with ease. Prithivi streamlines the process of setting up, running, and analyzing RBFE calculations and making physical simulations accessible to a broader range of scientists.
A Brief Introduction to Prithvi
Prithvi™ is our AI-powered platform to accelerate small molecule drug discovery with scientific foundation models. Prithvi™ has the ability to
- Perform structural analysis of targets and identify potential binding sites for a more targeted design process.
- Build models based on early assay or patent data for use in large-scale virtual screens.
- Construct active learning pipelines to identify and confirm high-quality hits.
- Guide retrosynthetic analysis.
- Suggest modifications to increase the potency, selectivity, and safety of hits.
To learn more about Prithvi™, check out our earlier blog post. In this blog post, we will focus on using Prithvi™ to perform Relative Binding Free Energy calculations for protein-ligand systems.
Running No-Code RBFE Calculations
As a case study, we leverage Prithvi™ for computing binding free energies of five ligands in conjunction with the TYK2 protein. Subsequently, we compare these outcomes with well-established experimental data to evaluate our results. Under the hood, we use OpenFE to perform RBFE simulations.
Simulation Setup
The dataset needed to run RBFE simulations is obtained from Open Force Fields’ openforcefield/protein-ligand-benchmark repository.
Protein - TYK2
We selected TYK2 protein due to its practical advantages for computational studies; notably TYK2 demonstrates easy convergence. Additionally, experimental data is available to validate our results.
Ligands
For the purpose of this experiment, we focus on 5 Ligands: ejm_48, ejm_31, ejm_46, jmc_28, and jmc_23 (displayed below). To mitigate the cost associated with RBFE calculations, we've constrained the ligand count to 5, consequently limiting the number of “edges” in our calculation to 4 (see the next section for details.)
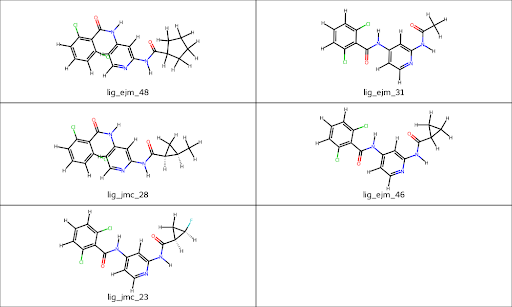
Prithvi™ aligns ligands and proteins, with ligand coordinates prepared in reference to the protein. Both protein and ligands are protonated to a single tautomeric state, following established practice in the field.
Reference Experimental Data
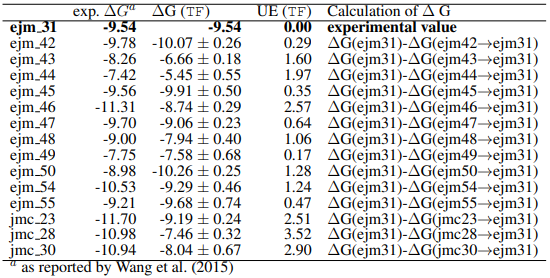
This datasheet provides experimental data for assessing benchmark results, with Table 1.1 presenting pertinent values specifically for the TYK2 System.
Planning the Simulations
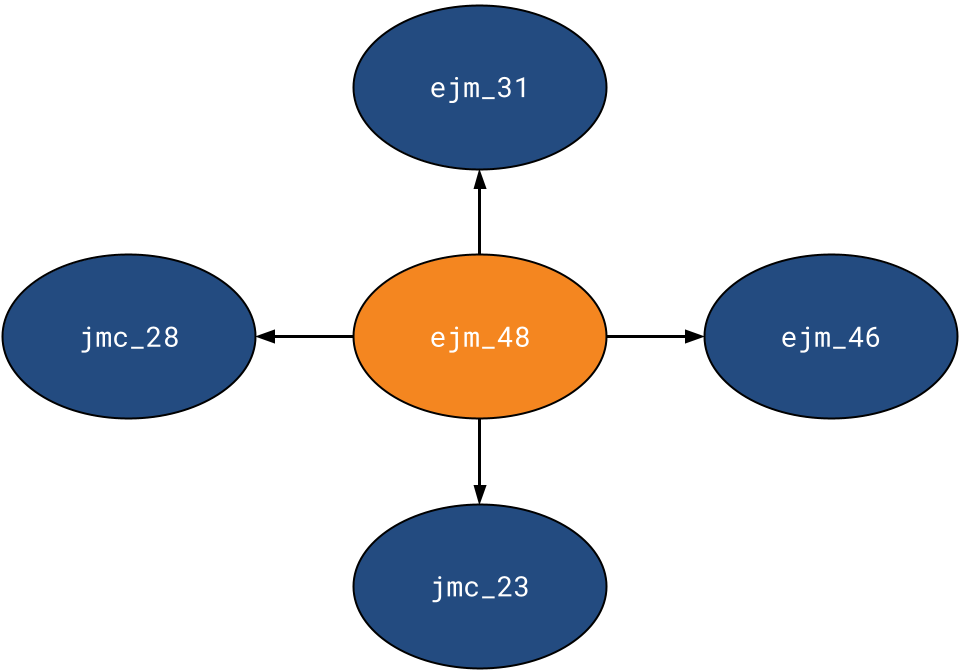
A singular RBFE calculation involves the protein and two ligands: the initial ligand of the transformation and the concluding ligand. These transformations, or perturbations, are represented as edges in a perturbation network— a directed graph where each edge signifies a transformation requiring RBFE computation. Various perturbation networks exist, including minimum spanning, radial, and maximal. Currently, Prithvi supports radial perturbation networks. A radial perturbation network resembles a star graph, where one node has a degree N, while all other nodes have a degree of 1. Prithvi supports maximal and minimal spanning networks as well.
In this simulation, we created a Radial Perturbation Network, positioning ejm_48 as the central node. The network comprised a total of 4 edges.
Running the Simulations
Employing the default settings extracted directly from openFE's RelativeHybridTopologyProtocol, we made a few adjustments. To reduce simulation times, we reduced the number of replicas from 11 to 5. Additionally, we set the lambda_windows to 5 to match the n_replicas.

Each edge in the process took approximately a day for completion, and we executed these computations on an AWS GPU node featuring the following specifications:
- Memory: 16 GB
- No. of GPUs: 1
- Graphics Memory: 16 GB
- GPU: Nvidia T4 Tensor Core
Results
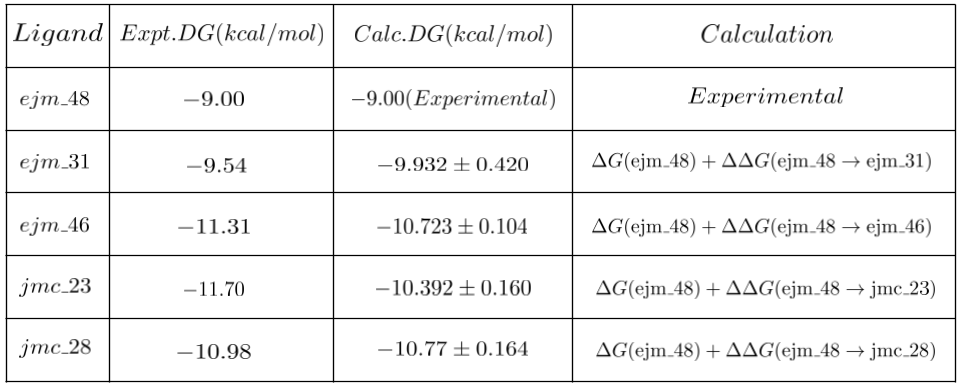
We observe that the computed binding free energy values for ejm_31 and jmc_28 closely align with the actual experimental values. In contrast, the binding free energies values for ejm_46 and jmc_23 exhibit some deviation from the experimental results, possibly due to lack of convergence. The cost of running these simulations amounted to $50, which is $12.5 per edge. These calculations were done on a dedicated GPU Node. Costs would be lower (at the cost of additional error handling) when we use SPOT compute instances.
Additionally, we conducted a singular edge simulation, maintaining default values for both n_replicas and lambda_windows, each set to 11. This simulation extended over a period of approximately three days, yielding the subsequent results.
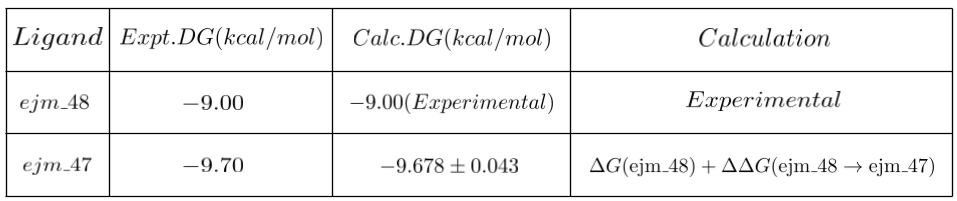
The computed binding free energy value for ejm_47 closely aligns with the reported experimental value. We believe employing default settings for the mentioned edges could have led to improved results for all other ligands. Nevertheless, considering the substantial computational expenses associated with these simulations, we opted for a reduced number of replicas to mitigate costs.
Conclusion and Takeaways
Prithvi's no-code platform enables scientists and researchers to perform RBFE calculation and analyze results without having to write any custom scripts. Our case study demonstrates how Prithvi™ can accurately compute relative binding free energies (RBFE) for protein-ligand systems that align well with experimental values for certain ligands. The computations, costing only $50 for five ligands, highlight Prithvi's cost-effectiveness. Prithvi™ is highly scalable and can be used to perform RBFE cost-effectively at large scales for your discovery needs.
About Deep Forest Sciences
Deep Forest Sciences’ no-code AI Prithvi™ toolchain accelerates small-molecule drug discovery efforts. Deep Forest Sciences also leads the development of open-source DeepChem framework, and emphasizes supporting open source and open science as fundamental parts of our mission and values. Partner with us to apply our foundational no-code AI technology to hard real-world problems in small molecule drug discovery.
Email us at partnerships@deepforestsci.com to learn more!