The Chemist's AI Co-Pilot: Introducing DeepRetro for Collaborative Retrosynthesis
Deep Forest Sciences
Shreyas Vinaya Sathyanarayana, Bharath Ramsundar
09.27.2025
The design of efficient synthetic routes for organic compounds is foundational to progress across the chemical sciences, from drug discovery to materials design. This process, known as retrosynthesis, involves logically deconstructing a complex target molecule into simpler, commercially available precursors. While conceptually elegant, navigating the vast search space of possible reactions is a formidable task that has long been a grand challenge for both chemistry and artificial intelligence.
Existing computer-aided synthesis planning (CASP) tools have shown promise but are often constrained by the reaction templates in their databases, limiting their ability to devise novel pathways. To address these limitations, we introduce DeepRetro, an open-source framework designed to facilitate a powerful, collaborative partnership between chemists and artificial intelligence.
The DeepRetro Architecture: An Iterative, Hybrid Approach
DeepRetro is a hybrid framework that integrates Large Language Models (LLMs), traditional retrosynthesis engines, and expert human feedback in an iterative design loop. Instead of attempting to generate a complete synthetic route in a single pass, the system employs a step-wise refinement process:
- Initially, a traditional template-based tool attempts to find a retrosynthetic disconnection.
- If this tool fails to find a solution, the framework queries an LLM (such as Anthropic's Claude series or DeepSeek R1) to propose a single-step disconnection.
- This LLM-generated suggestion is not accepted blindly. It undergoes a series of rigorous validation steps - including checks for chemical validity, stability, and potential "hallucinations" - to ensure chemical plausibility.
- Validated precursors are then recursively fed back into the planning loop, allowing for dynamic course correction and the systematic construction of a multi-step pathway.
- This iterative methodology combines the generative flexibility of LLMs with the precision of template-based methods, all while enforcing chemical rigor at each step.
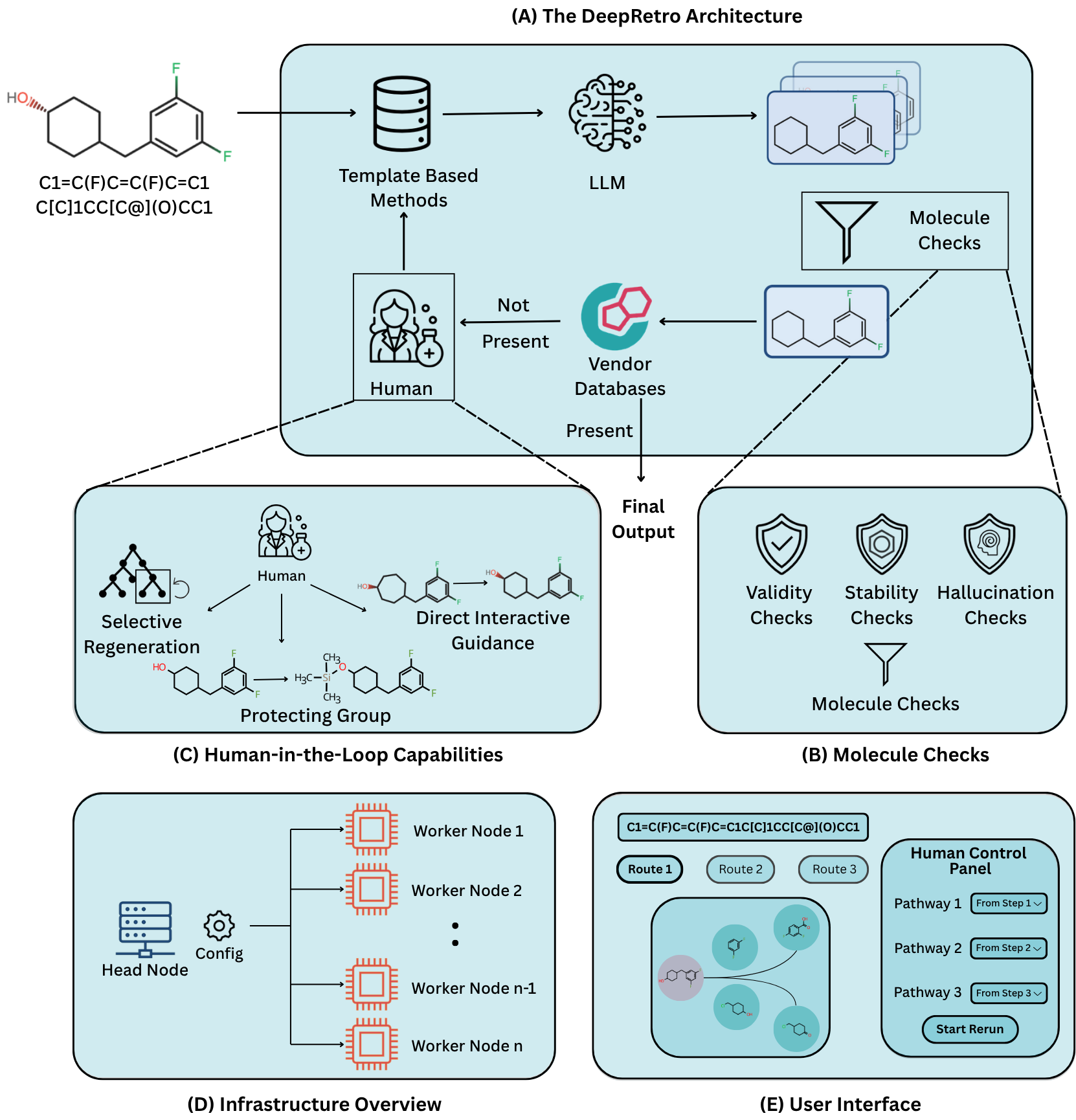
Human-in-the-Loop: A Partnership for Discovery
A core design principle of DeepRetro is to empower chemists by incorporating several human-in-the-loop (HITL) capabilities, recognizing that the combination of machine-generated routes and expert intuition leads to the most practical outcomes. Through an interactive graphical user interface (GUI), domain experts can visualize and intervene in the retrosynthetic planning process in real-time.
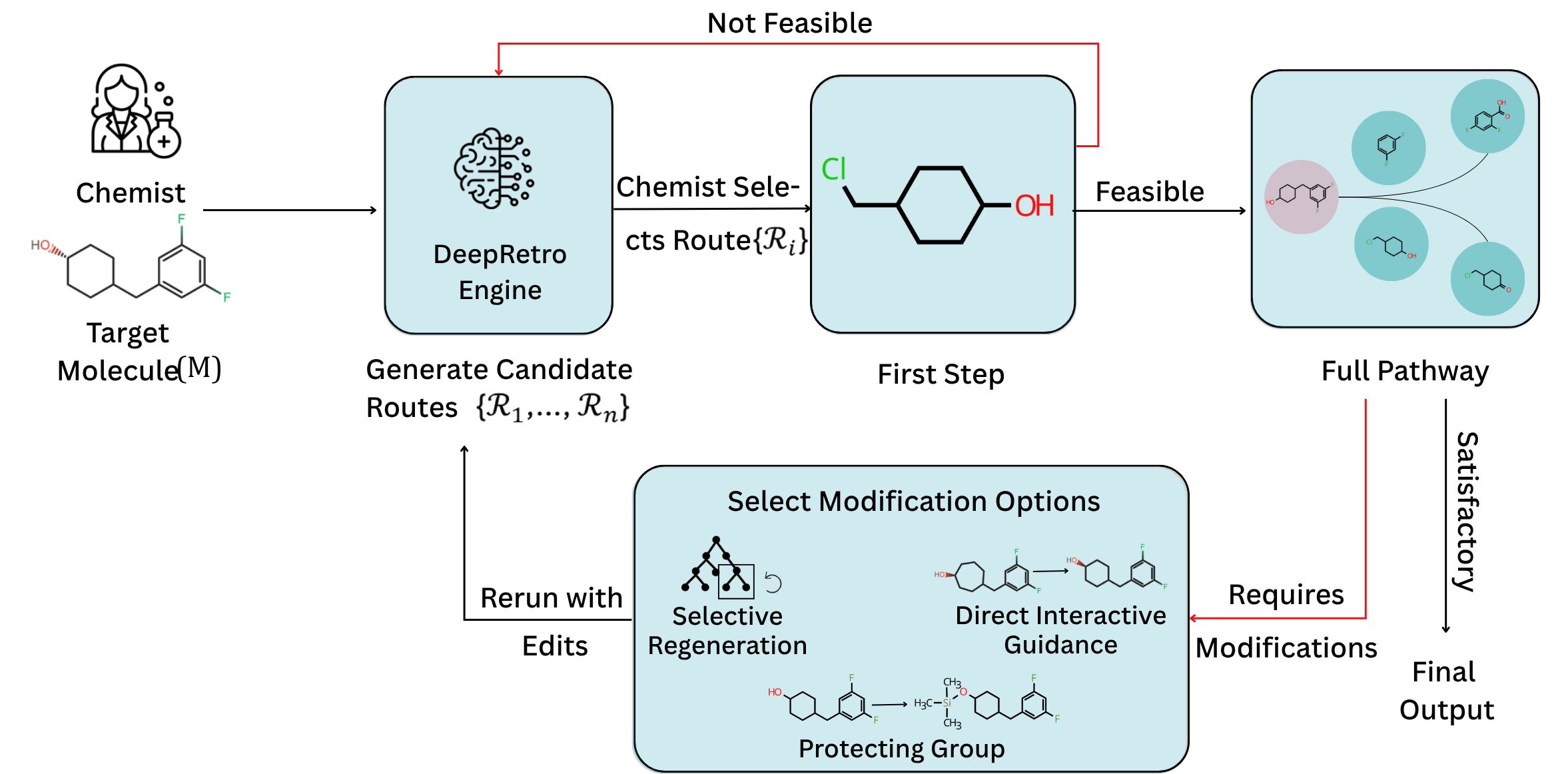
Key collaboration features include:
- Selective Pathway Regeneration: Chemists can identify a suboptimal step within a proposed route and instruct the system to regenerate only that segment, allowing for targeted refinement without discarding the rest of the pathway.
- Direct Interactive Guidance: The interface allows chemists to directly edit molecular structures (e.g., SMILES strings) to correct minor errors or LLM-induced hallucinations, ensuring chemical accuracy in subsequent steps.
- Strategic Protecting Group Manipulation: Users can designate reaction sites to introduce, modify, or remove protecting groups - a critical aspect of practical multi-step synthesis.
This collaborative framework proved essential for navigating the synthesis of complex natural products.
Validation and Performance
We evaluated DeepRetro on standard retrosynthesis benchmarks and complex case studies. In automated mode, the framework achieves state-of-the-art performance, with the Claude 4 Opus configuration successfully solving 96.3% (183/190) of targets in the USPTO-190 multi-step benchmark.
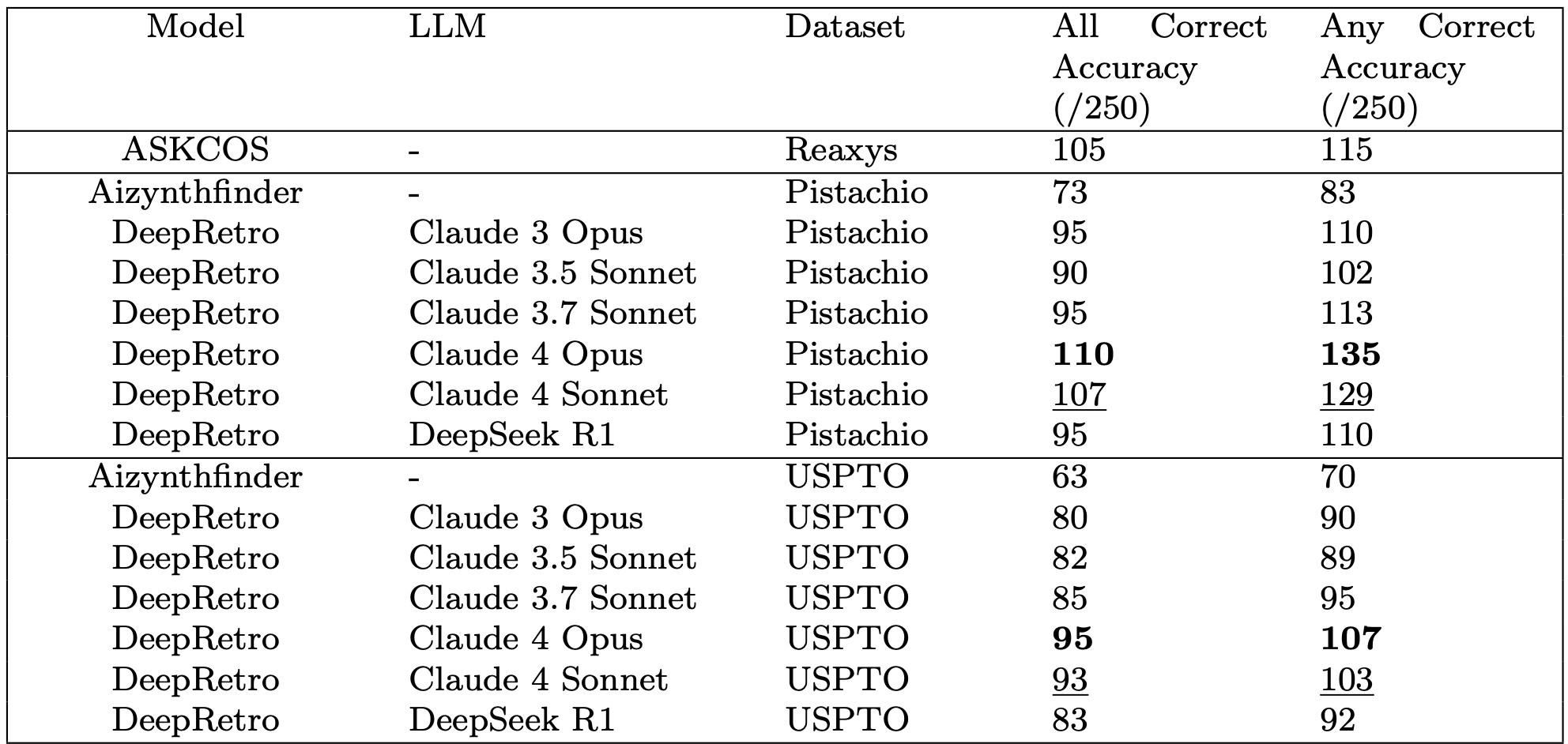
Case Studies: From Strong Performance to Novel Science
The true potential of DeepRetro is demonstrated in its collaborative use for complex natural products. For two of the five molecules in our case studies (Ohuamine C and a Tetracyclic Azepine Derivative), the DeepRetro framework enabled the discovery of novel synthetic pathways not previously reported in the literature. This is actual novel science, made possible by the synergy between an expert chemist and an LLM.
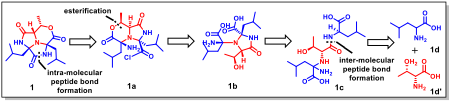
Novel Pathway for Ohauamine C
For the complex natural product Ohauamine C, DeepRetro proposed a novel strategy employing an unprecedented early-stage esterification. This key step pre-organizes the molecular conformation, enabling a more efficient macrocyclization later in the synthesis - a significant departure from conventional methods.
Novel Pathway for a Tetracyclic Azepine Derivative
For this tetracyclic derivative, the framework identified a novel disconnection at the tertiary amine, allowing the molecular scaffold to be constructed from simpler fragments. This convergent approach, which leverages an amine-epoxide ring-opening reaction, represents a conceptual shift from existing literature strategies for this class of molecules.
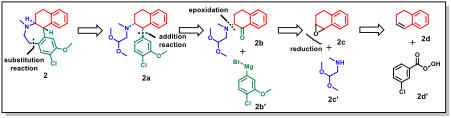
These discoveries illustrate DeepRetro's capacity to not only find solutions but to facilitate genuine scientific discovery.
A Model for Future Scientific Collaboration
DeepRetro serves as a working model for how LLMs can be effectively integrated into scientific discovery pipelines, a field rapidly advancing with recent announcements from teams at OpenAI on accelerating life sciences and Google DeepMind on single-cell analysis. While these exciting efforts show the power of LLMs for analyzing complex biological data, DeepRetro demonstrates a concrete application of LLMs for generative science - creating novel, plausible pathways in chemistry.
We have shown that this approach can lead to the discovery of new science. However, due to the high rate of chemically implausible "hallucinations" from current general-purpose LLMs, the system still relies critically on human-in-the-loop feedback for complex targets. This partnership, where the LLM provides creative suggestions and the human expert provides validation and guidance, is currently the most effective path to discovery.
Despite this limitation, we see a clear pathway to a future of fully automatic LLM-driven discovery. As LLMs become more chemically fluent and our validation frameworks grow more sophisticated, the need for intervention will decrease. DeepRetro's iterative, validated architecture lays the groundwork for that future.
To enable the broader scientific community to benefit from and build upon our work, we have made the entire DeepRetro framework open-source. We believe this tool will empower chemists to tackle increasingly ambitious synthetic targets and accelerate progress in drug discovery, materials design, and beyond.
Explore the framework and the paper here:
- GitHub repository: https://github.com/deepforestsci/DeepRetro
- Read the full paper: [DeepRetro: Retrosynthetic Pathway Discovery using Iterative LLM Reasoning](https://arxiv.org/abs/2507.07060)
References
[1] https://openai.com/index/accelerating-life-sciences-research-with-retro-biosciences/
[2] https://research.google/blog/teaching-machines-the-language-of-biology-scaling-large-language-models-for-next-generation-single-cell-analysis/
About Deep Forest Sciences
Deep Forest Sciences’ no-code AI Prithvi™ toolchain accelerates small-molecule drug discovery efforts. Deep Forest Sciences also leads the development of open-source DeepChem framework, and emphasizes supporting open source and open science as fundamental parts of our mission and values. Partner with us to apply our foundational no-code AI technology to hard real-world problems in small molecule drug discovery.
Email us at partnerships@deepforestsci.com to learn more!