No-Code Molecular Dynamics with Prithvi
Deep Forest Sciences
Shashank SM, Jose Antonio Siguenza, Rida Irfan, Bharath Ramsundar
06.25.2024
A Brief Overview of Molecular Dynamics
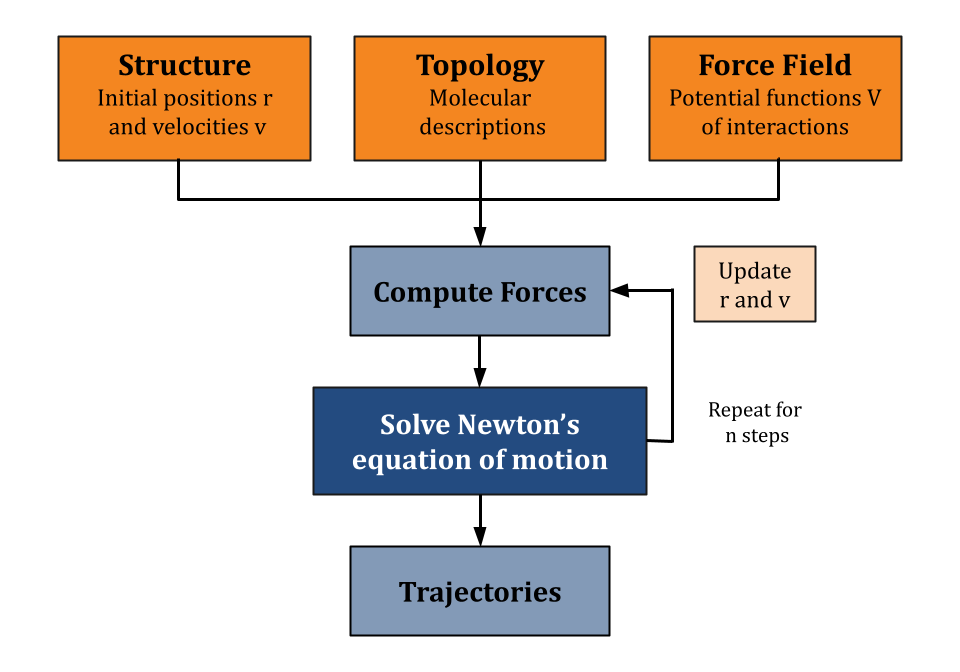
Molecular Dynamics (MD) is a computational method used to study the physical movements of atoms and molecules, such as proteins and nucleic acids, at the atomic level. The technique simulates the movement of atoms over time based on the interactions and forces between them, following Newton's laws of motion.
Molecular systems are complex and can consist of vast number of atoms and molecules. Analytically predicting the properties and behaviors of these systems under various conditions is extremely challenging. MD simulations avoid analytical solutions by numerically integrating Newton's equations of motion to model the trajectories of individual atoms. At each step, the method calculates the forces exerted on each atom by all other atoms in the system and iteratively updates their positions and velocities over time. Molecular dynamics is considerably more efficient, and can handle larger systems and longer simulation times, than quantum mechanical simulation methods. At the same time, it is far slower than coarse-grained models of pathways, cells or tissues from systems biology.
MD simulations use force fields, mathematical models of the potential energy surfaces of molecules. Forces are computed by computing the negative gradient of the potential energy. Force fields include parameters for bond lengths, angles, and non-bonded interactions such as van der Waals forces and electrostatic interactions. The simulation process involves dividing time into small steps (typically at the femtosecond scale), during which the forces on each atom are computed and their positions and velocities are updated accordingly. This iterative approach allows researchers to capture the time-dependent evolution of molecular systems.

Molecular dynamics simulations have recently found greater use in drug discovery. For example, a recent article demonstrates how MD simulations provide a powerful complement to experimental methods. MD simulations can help identify the binding modes of potential drugs with their target proteins. Additionally, MD simulations provide information on how proteins change shape and interact with other molecules in their environment. This can be used to better understand the function of proteins and design drugs that modulate their activity. MD simulations can aid in virtual screening by helping to identify alternative binding poses. Experimental techniques like NMR spectroscopy and fluorescence quenching which validate the predictions of MD simulation remain critical, but simulations are slowly becoming useful predictive tools[1].
Advances in computational power and the availability of open source software are powerful trends driving adoption of molecular dynamics. However a number of practical challenges remain to broad adoption. Several research efforts have integrated machine learning with MD simulations to improve accuracy of simulations and enable the prediction of unsampled molecular configurations [2]. In addition, MD simulations remain computationally expensive and generate vast amounts of data, making it challenging to analyze and identify key molecular behaviors.
No-Code Molecular Dynamics with Prithvi
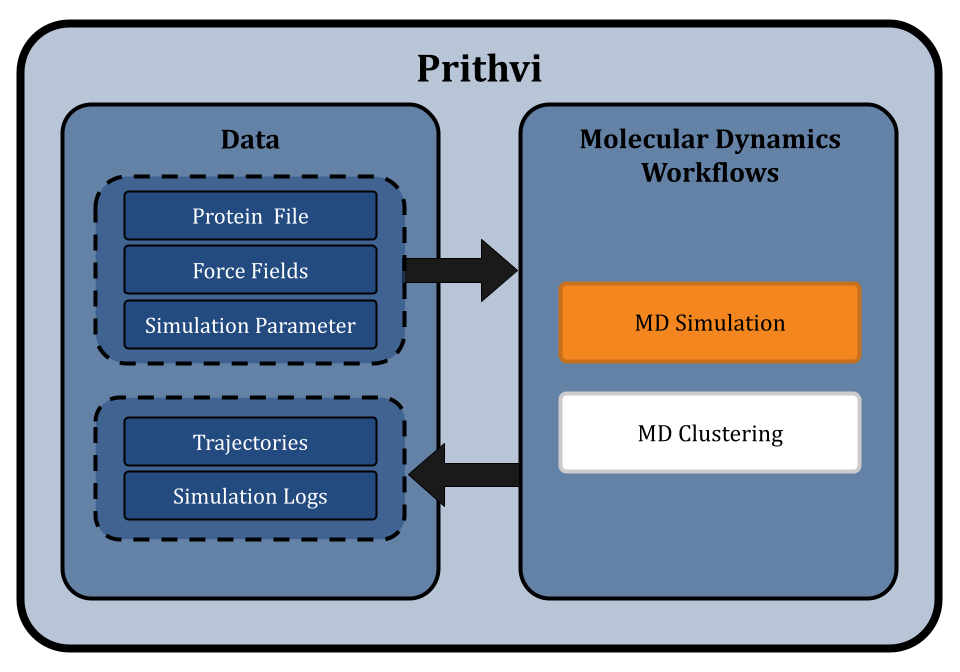
Effectively using molecular dynamics simulations involves complex and time-consuming calculations and often requires advanced computational skills. Prithvi™ provides a no-code platform for biologists and chemists to run molecular dynamics workflows to simulate and analyze molecular interactions without needing such expertise. Prithvi™ simplifies the entire process of MD simulations and allows the users to upload molecular data, select suitable force fields, control temperature and pressure, and configure other parameters as they require.
Brief Introduction to Prithvi
Prithvi™ is our AI-powered platform to accelerate small molecule drug discovery with scientific foundation models. Prithvi™ has the ability to
- Perform structural analysis of targets and identify potential binding sites for a more targeted design process.
- Build models based on early assay or patent data for use in large-scale virtual screens.
- Construct active learning pipelines to identify and confirm high-quality hits.
- Guide retrosynthetic analysis.
- Suggest modifications to increase the potency, selectivity, and safety of hits.
To learn more about Prithvi™, check out our earlier blog post. In this blog post, we will focus on using Prithvi™ to run protein-based MD simulations.
No-Code Molecular Dynamics
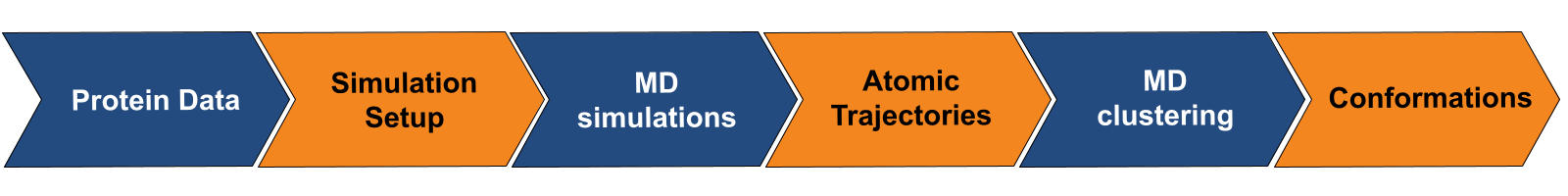
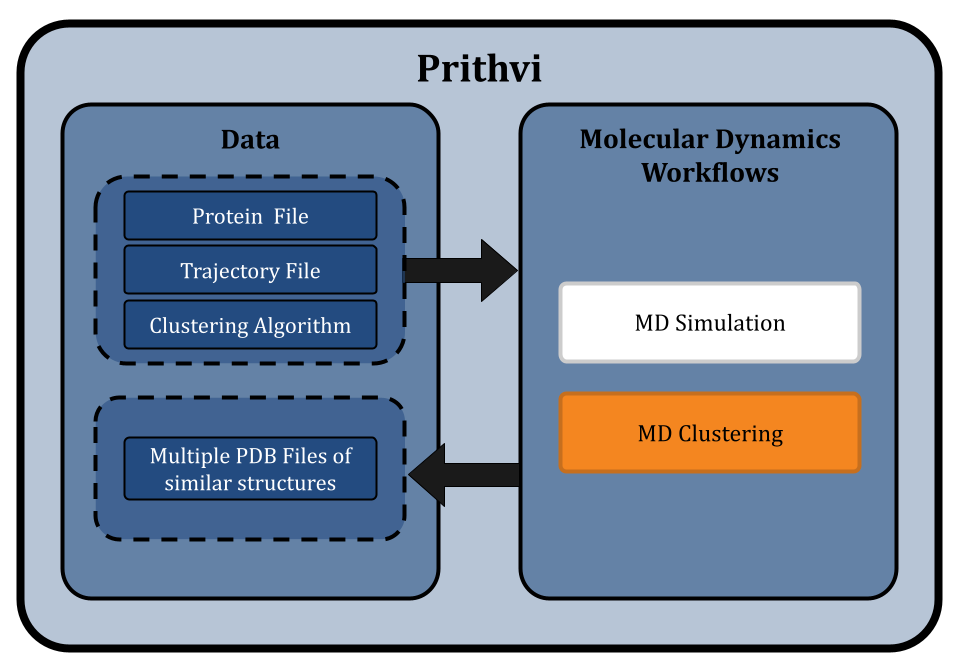
Prithvi™ offers a no-code platform for running Molecular Dynamics (MD) simulations, making complex molecular analyses accessible without extensive coding. Prithvi’s molecular dynamics workflow allows scientists to specify and run MD simulations directly through its web UI without having to write custom scripts or set up high performance computing (HPC) environments. Prithvi’s MD workflow involves the following steps
- Uploading and cleaning PDB
- MD simulations
- MD clustering
Uploading and Cleaning PDB
To start an MD simulation on Prithvi™, users upload a Protein Data Bank (PDB) file containing the three-dimensional structure of the molecule(s) to simulate. This file defines the initial positions of all atoms in the biomolecular system. PDB files may contain errors that can cause the MD simulations to become unstable and give unrealistic results. Cleaning involves tasks like removing water molecules, adding missing atoms, fixing structural issues, etc. to prepare the structure for MD simulation.
Running Molecular Dynamics Simulations
Under the hood, Prithvi™ makes use of OpenMM. Force fields such as such as, amber14-all, amber14/tip3pfb and charmm36/tip5pew, provide mathematical models that describe potential energy surfaces and the forces acting on atoms within molecular systems. Prithvi™ supports several force fields, which provide different methods of modeling interactions between atoms.
Prithvi™ uses the LangevinMiddleIntegrator by default but also supports other integrators. Prithvi™ enables users to control the temperature and pressure of the system to simulate different environmental conditions, such as a temperature of 300 K (27℃) to mimic biological environments. This control allows researchers to study how molecular interactions change under various conditions.
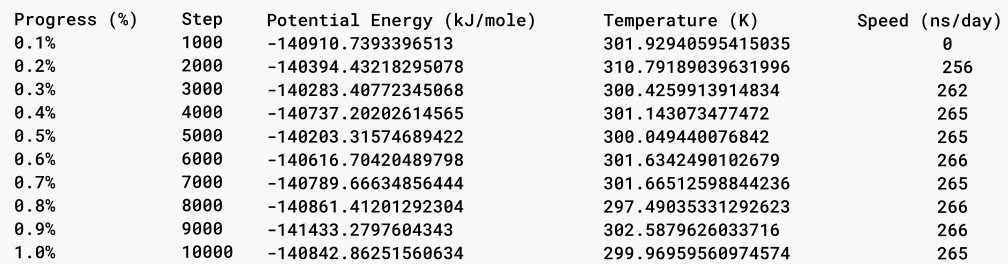
Simulation setup allows for several additional parameters, such as step size, simulation duration, and reporting intervals, to be set. For example, a typical simulation might run for 0.5 nanoseconds with a step size of 0.0005 picoseconds and log results at regular intervals. This detailed control over simulation parameters allows users to tailor their simulations to specific research needs.

Prithvi allows users to use GPU nodes for simulations and provides for easy use of both dedicated and spot instances.
Once the simulation is complete, Prithvi™ generates trajectory files that store the time-dependent coordinates and velocities of the atoms in DCD format. These files provide a comprehensive record of the simulated molecular dynamics, allowing researchers to visualize and analyze the behavior of the molecules over time. Additionally, Prithvi™ stores produced log files detailing the various parameters and conditions throughout the simulation.
Molecular Dynamics Clustering
MD simulations generate extensive trajectories of atomic positions over time, showing the various conformational states. These resulting trajectories contain large amounts of data. To make sense of this data, clustering techniques are employed to group similar conformations together which allows researchers to identify and analyze significant structural states within the molecular system.
Prithvi’s no-code MD workflow implements several clustering algorithms, like affinity propagation, DBSCAN and k-means, to efficiently analyze MD trajectory data generated from MD simulations to group similar molecular structures and form clusters of conformations.
To configure the affinity propagation algorithm, users set parameters such as damping, maximum iterations, convergence iterations, and preference.

Prithvi™ runs the selected clustering algorithm and generates several PDB files, each representing a distinct cluster of similar molecular structures identified in the trajectory file. These clusters provide an easy method to identify distinct conformational states, possibly corresponding to different biological function, that the molecule can adopt.
Prithvi provides a molecular docking framework for the users to predict protein-ligand interactions and design molecules that bind to the structures generated by the MD workflow This is done using no-code tools for pocket finding, pose generation and scoring. For detailed information on Prithvi’s no-code molecular docking capabilities, see our earlier blog post.
Conclusion and Takeaways
Molecular Dynamics has been used widely to study the structure and dynamics of macromolecules like proteins. MD simulations can be particularly valuable in lead optimization where they can help refine ligand interactions, predict binding pocket rearrangements, and improve ligand binding poses. Unlike traditional docking methods, MD simulations offer the potential for more accurate predictions of ligand binding affinities [1]. However, this process is computationally intensive and requires sophisticated knowledge. Prithvi™ provides a no-code molecular dynamics workflow for the researchers to easily run MD simulations and clustering for understanding and analyzing molecular behavior and interactions, without requiring advanced computational expertise.
MD simulations facilitate virtual screening by enabling systematic study of the flexibility of binding pockets, and can help identify a broader range of potential binders compared to docking with a single protein structure. MD simulations also help understand conformational changes that influence specific signaling patterns, needed for designing drugs with precise therapeutic effects [1]. Using no-code primitives, Prithvi™ streamlines the entire process of running molecular dynamics simulation by allowing users to upload molecular data, select suitable force fields, and control environmental conditions as they require. In future blog posts, we will discuss more advanced applications of this technology, such as showing how Prithvi™ can be used to facilitate the design of allosteric inhibitors.
References
[1] Hollingsworth, S. A., & Dror, R. O. (2018). Molecular dynamics simulation for all. Neuron, 99(6), 1129–1143. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6209097/
[2] Wang, Y., Ribeiro, J. M. L., & Tiwary, P. (2019). Machine learning approaches for analyzing and enhancing molecular dynamics simulations. arXiv preprint arXiv:1909.11748. https://arxiv.org/abs/1909.11748.
About Deep Forest Sciences
Deep Forest Sciences’ no-code AI Prithvi™ toolchain accelerates small-molecule drug discovery efforts. Deep Forest Sciences also leads the development of open-source DeepChem framework, and emphasizes supporting open source and open science as fundamental parts of our mission and values. Partner with us to apply our foundational no-code AI technology to hard real-world problems in small molecule drug discovery.
Email us at partnerships@deepforestsci.com to learn more!